
 Get More Help in Utah
Get More Help in UtahSpinal Muscular Atrophy
Overview

Other Names & Coding
G12.0, Spinal muscular atrophy, type I; Werdnig-Hoffman
G12.1, Other inherited spinal muscular atrophy
G12.9, Spinal muscular atrophy, unspecified
ICD-10 for Spinal Muscular Atrophy and Related Syndromes (icd10data.com) provides coding details.
Prevalence
Genetics
The severity of SMA is dependent on a second gene, SMN2, also on 5q13. SMN2 is the result of an ancestral duplication and is identical to SMN1 except for the presence of a missense mutation on exon 7 that disrupts a splicing enhancer but does not change the predicted protein sequence. This mutation results in mis-splicing of exon 7 and an inactive transcript; however, at a low rate, normal splicing occurs, providing a small amount of normal transcript and normal SMN protein. Severity of clinical symptoms is correlated with the number of copies of SMN2 since patients with more copies of SMN2 produce more normal SMN protein. While not perfectly correlated, most SMA type I patients have 1 or 2 SNM2 copies, and most patients with SMA type III have 3 or more copies. Treatments targeting up-regulation of expression of normal transcript from SMN2 have proven successful clinically, resulting in approval of nusinersen in 2016 and risdiplam in 2020.
With the addition of SMA to the recommended uniform screening panel, newborn screening programs for SMA are rapidly coming online in many states. Early initiation of treatment leads to far better outcomes, especially if treatment is started before the onset of symptoms and before significant loss of motor neurons becomes permanent. In most cases, newborn screening will identify homozygous deletion of SMN1 and will not identify carrier status or SMN2 copy number. Confirmatory testing, including SMN2 copy number after a positive newborn screen, is essential. [Baker: 2019] [Ke: 2019]. As newborn screening only detects infants with SMN1 deletions, about 95% of all cases of SMA but will miss those infants with one SMN1 deletion and one point mutation on the second copy.
Prognosis
Individuals with type II and type III SMA can have a normal life span but experience significant disability and respiratory or other complications. Anticipatory care that includes careful attention to respiratory, nutritional, and orthopedic issues can significantly prolong survival.
The availability of treatments, such as nusinersen, risdiplam, and onasemnogene abeparvovec-xioi (detailed in the Management section, below), has greatly changed the natural history and progression of SMA, especially when treatment is initiated early. [Ross: 2019] Although long-term data over a decade or more are not yet available, in many cases, infants who would not have lived past 2 years old are surviving longer and maintaining relatively good health. [De: 2019]
Practice Guidelines
The standard of care guidelines (below) represents expert consensus but lack
well-designed clinical trials to validate recommendations. It is expected that
the standards of care will be updated in the near future to reflect new data
from approved treatments and ongoing studies.
Kirschner J, Butoianu N, Goemans N, Haberlova J, Kostera-Pruszczyk A, Mercuri E, van der Pol WL, Quijano-Roy S, Sejersen T,
Tizzano EF, Ziegler A, Servais L, Muntoni F.
European ad-hoc consensus statement on gene replacement therapy for spinal muscular atrophy.
Eur J Paediatr Neurol.
2020;28:38-43.
PubMed abstract / Full Text
Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J, Main M, Mazzone ES, Vitale M, Snyder B, Quijano-Roy S, Bertini E, Davis
RH, Meyer OH, Simonds AK, Schroth MK, Graham RJ, Kirschner J, Iannaccone ST, Crawford TO, Woods S, Qian Y, Sejersen T.
Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and
nutritional care.
Neuromuscul Disord.
2018;28(2):103-115.
PubMed abstract
Finkel RS, Mercuri E, Meyer OH, Simonds AK, Schroth MK, Graham RJ, Kirschner J, Iannaccone ST, Crawford TO, Woods S, Muntoni
F, Wirth B, Montes J, Main M, Mazzone ES, Vitale M, Snyder B, Quijano-Roy S, Bertini E, Davis RH, Qian Y, Sejersen T.
Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations;
other organ systems; and ethics.
Neuromuscul Disord.
2018;28(3):197-207.
PubMed abstract
Michelson D, Ciafaloni E, Ashwal S, Lewis E, Narayanaswami P, Oskoui M, Armstrong MJ.
Evidence in focus: Nusinersen use in spinal muscular atrophy: Report of the Guideline Development, Dissemination, and Implementation
Subcommittee of the American Academy of Neurology.
Neurology.
2018;91(20):923-933.
PubMed abstract
Roles of the Medical Home
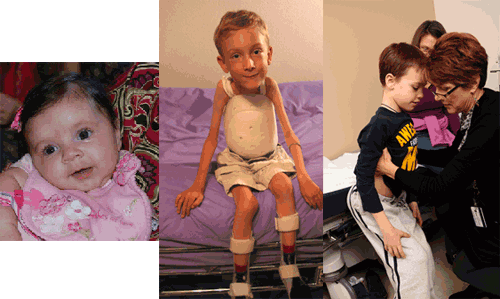
Clinical Assessment
Overview
Pearls & Alerts for Assessment
Urgent referral when positive newborn screen for SMAA positive newborn screen for SMA should prompt urgent referral to a neuromuscular center where treatments can be coordinated quickly, before onset of symptoms. Outcomes for pre-symptomatic treatment are far superior to treatment at later stages. In one study of 25 infants with 2 or 3 copies of SMN2, treated pre-symptomatically with nusinersen, 22 of 25 reached independent ambulation. [De: 2019] For pre-symptomatic infants with SMA, symptoms can arise in the first weeks of life and time is critical. Delay of treatment even for a few days can result in permanent disability. [Butterfield: 2021]
Newborn screening will not pick up all cases of SMAIf SMA is suspected even in a child with negative newborn screening, further testing (SMN1 sequencing) is needed as newborn screening only identifies SMN1 deletion and 5% of patients are expected to have other mutations in the SMN1 gene. Children with one SMN1 deletion and a one point mutation on the second copy of SMN1 would have a negative screen.
Address possibility of respiratory failureInfants with type I SMA are susceptible to respiratory failure due to infection or aspiration. Addressing this possibility early with the family and implementing proactive measures (e.g., non-invasive ventilation, cough assist, and G-tube placement) can prevent emergency situations. Frequent pneumonias or respiratory illnesses may signal impending respiratory failure or aspiration.
Screening
For the Condition
Of Family Members
Presentations
- Type I SMA (Werdnig-Hoffman) presents in the first few weeks of life. Children never learn to sit or walk and have severe respiratory and swallowing problems, including difficulty handling oral secretions and a significantly shortened life span. Other features include poor head control, a bell-shaped chest, weak cry and cough, tongue atrophy and fasciculation, and paradoxical breathing.
- Type II SMA presents later in the first year of life or up to about 2 years of age. Children usually learn to sit but do not walk. Fine tremor-like movements in the hands and fingers may be noted from early in the course. As weakness progresses, children often have respiratory and swallowing problems, including difficulty gaining weight due to bulbar muscle weakness, weak cough, and nighttime hypoventilation. Swallowing problems and difficulties opening the jaw widely have been shown to contribute to malnutrition. [Messina: 2008] Joint contractures and scoliosis develop over time in nearly all affected children and warrant proactive intervention.
- Type III SMA (Kugelberg-Welander) is much more variable in onset than types I and II, but it usually presents in childhood or early adolescence. Children can sit and walk (although some may lose this ability over time). As with type II SMA, children with type III SMA may have fine tremor-like movements in the hands and fingers. Quadriceps atrophy may be pronounced. Children with type III SMA may have fewer problems with respiratory function and swallowing than children with type I or II SMA. Scoliosis, contractures, and joint pain are often noted in older children.
- Type 0 SMA presents before birth with decreased fetal movement noted around 30 weeks of gestation. Newborns are severely hypotonic at birth with congenital contractures, swallowing problems, and respiratory failure.
- Type IV SMA presents in adults and will not be discussed further here.
Diagnostic Criteria
Identification of the copy number of the SMN2 gene assists in prognosis and is a standard feature of genetic testing for SMA. While most patients will have the common deletion of exon 7, about 5% of children who present with SMA have negative genetic testing due to presence of a deletion on 1 allele and a point mutation on the other. Clinical suspicion, more detailed gene testing, and electromyography (EMG) play important roles in diagnosis in these cases.
Clinical Classification
- Children with type I SMA, which is the most common form, present before 6 months of age with generalized muscle weakness and progressive respiratory compromise. These children never achieve independent sitting.
- Children with type II SMA present between 6 months and 1 year of age with weakness and motor regression. Children with type II SMA sit independently but do not walk.
- Onset for type III SMA is variable from early childhood to adolescence. Children with type III SMA achieve independent ambulation, although they do not necessarily retain ambulation throughout life.
Differential Diagnosis
Infant botulism occurs in children up to 12 months of age. Symptoms start with constipation in a previously normal baby and are followed by decreased facial expression, poor swallowing, a weak cry, and decreased movement. Over time, the course is more acute than SMA. Diagnosis is made by recognition of clinical features and demonstration of botulinum toxin in the infant's stool. EMG can help to exclude other diagnoses.
Neuropathies are a wide spectrum of syndromes with various time courses. Sensory nerves are usually involved. Family history is often positive (e.g., Charcot-Marie-Tooth disease or hereditary motor sensory neuropathy). Acquired polyneuropathies, such as Guillain-Barre syndrome, have a rapid onset over a few days to a week. These are exceedingly rare in children under 2 years old. Diagnosis is confirmed by EMG and/or tests showing elevated cerebrospinal fluid (CSF) protein.
Metabolic myopathy (e.g., mitochondrial myopathy, Pompe disease) is much less common than SMA, but early features can be clinically similar. Infants with Pompe disease have severe progressive cardiac dysfunction not seen in SMA.
Duchenne muscular dystrophy occurs in boys only. In this condition, calves are large, creatine kinase (CK) is extremely high, and developmental delay is often present. It is diagnosed by genetic testing for mutations in the DMD gene.
X-linked SMA and SMA with respiratory distress (SMARD) may appear clinically similar to SMA, but they have different genetic etiologies and are therefore not currently amenable to treatment. SMARD1 presents as distal (not proximal) muscle weakness, with foot deformities as well as respiratory failure that often occurs suddenly. This condition is due to mutations in the IGHMBP2 gene on chromosome 11q13.3, which encodes the immunoglobulin micro-binding protein 2. [Kaindl: 2008] Infants with X-linked SMA may have a family history showing X-linked inheritance and will have negative SMN1 testing. An increasing number of new genes are being associated with these phenotypes and suspicion should prompt referral to a sub-specialty clinic for assessment.
Congenital myopathy presents with non-progressive weakness and is diagnosed by genetic testing and/or muscle biopsy.
History & Examination
Early recognition of weakness is critical to the initial evaluation. Assessment of weakness can be difficult in young children, but close attention to functional ability allows a good assessment of strength. Videos of children with and without weakness are available at the Child Muscle Weakness Organization and can be helpful in identifying subtle weakness.
Current & Past Medical History
Respiratory: Ask about breathing problems while awake, including while eating, for infants. Ask about history of pneumonia, strength of cough, reactive airway disease, recent pulmonary testing (oximetry), use of respiratory support (noninvasive respiratory support such as CPAP and BiPAP and nighttime or continuous ventilation), and use of a cough assist machine. Ask also about sleep, results of previous sleep studies, snoring as evidence of obstructive sleep apnea and hypoventilation, frequency of night waking, daytime sleepiness, and morning headaches. Check immunization history, including pneumococcal and flu vaccines.
Swallowing problems: Ask about coughing or choking while eating and drinking (especially thin fluids) and strength of cough. Ask if the family has a suction machine at home.
Musculoskeletal: Ask about any recent changes in muscle strength, fatigability, and functional abilities. Ask about fit and maintenance of the wheelchair, stander, or other equipment. Ask if the family has a license plate or placard for individuals with disabilities.
Orthopedic: Ask about orthotics use, joint pain, range of motion, and history of fractures.
HEENT: Ask about chronic ear infections.
Dental: Ask about dental history and the ability to open and close jaw.
Family History
Pregnancy/Perinatal History
Developmental & Educational Progress
Social & Family Functioning
Physical Exam
General
Growth Parameters
Wt | Ht | BMI: Due to feeding problems, infants with SMA may be underweight or overweight (particularly when using a G-tube to augment feedings). Careful attention to feeding and growth patterns is important. With decreased muscle mass, a relatively low weight for length may be ideal. Standard growth charts for children may be inappropriate.
Skin
HEENT/Oral
Check ability to open jaw and the size of tonsils. Check for bad breath, dental caries, and drooling that may be caused by swallowing problems.
Chest
The chest wall may appear bell-shaped. Listen to the lungs and observe for evidence of breathing difficulties. Check respiratory rate and look for paradoxical abdominal/chest wall movements. Observe cough or cry if possible. Consider spot oximetry, peak flow, and forced vital capacity measures. Measure chest circumference at the nipple line and follow growth. Chest and head circumference should be fairly equal in the first year. If the chest is not growing, the child is at a higher risk of respiratory compromise.
Testing
Laboratory Testing
Imaging
Genetic Testing
Although the number of SMN2 copies is not needed to diagnose SMA, guidelines recommend that this be obtained to allow assessment of the predicted SMA phenotype. In most cases, those with type I SMA have 2 copies, type II SMA have 3 copies, and type III SMA have 4 copies of SMN2. In addition, eligibility for medication trials for SMA may depend on SMN2 copy number. [Mercuri: 2018] See Testing for SMA (Genetic Testing Registry).
Other Testing
- Electromyography (EMG) can indicate probable SMA, in which case, genetic testing should follow.
- Pulmonary function tests and sleep studies are used to evaluate respiratory status as needed.
- Nutritional assessments are helpful for evaluation of children who are underweight or overweight.
- DEXA scans are performed if the child has a history of fractures or, as a baseline if the child has been immobile and confined to a wheelchair for an extended period, especially if the child is on valproic acid or proton pump inhibitors.
Specialty Collaborations & Other Services
Neuromuscular Clinics (see UT providers [3])
Medical Genetics (see UT providers [7])
Pediatric Pulmonology (see UT providers [3])
Pediatric Gastroenterology (see UT providers [2])
Pediatric Orthopedics (see UT providers [10])
Pediatric Physical Medicine & Rehabilitation (see UT providers [11])
Pediatric Otolaryngology (ENT) (see UT providers [10])
Treatment & Management
Pearls & Alerts for Treatment & Management
New specific treatments are available for SMA
Nusinersen (Spinraza) is now approved by the FDA for
individuals with all types of SMA. There is emerging data supporting
dramatic benefits even in severely affected patients, but long-term outcomes
are not known. Nusinersen has generally been well-tolerated. There is Class
III evidence that infants with homozygous mutations/deletions of
SMN1 have improved, ventilation-free survival at
age 24 months. Finally, there is evidence that in children aged 2-12
diagnosed after 6 months of age (SMA type II patients) nusinersen results in
greater improvement in motor function after 15 months of treatment.
Nusinersen is extremely expensive, and there are
many inequalities between children without insurance and children with
various types of insurance. It needs to be given by lumbar puncture and a
controlled environment and sedation are needed. Nusinersen is given by 4
loading doses (the first 3 at 14-day intervals and the 4th 30 days after the
third dose) and then 1 dose every 4 months indefinitely. Administration of
nusinersen may be particularly difficult in children who develop scoliosis
and in the setting of spine surgery. In addition to the price of nusinersen,
administration costs also need to be considered.
Onasemnogene abeparvovec-xioi
(Zolgensma) was approved by the FDA in2019 for children
under 2 years of age with SMA. Onasemnogene abeparvovec-xioi is administered
by IV as a one-time dose as soon as possible after diagnosis. To qualify
currently, children must be under 2 with bi-allelic mutations in the
survival motor neuron 1 (SMN1) gene and absent
antibody to the vector, AAV9. Children are treated with steroids one day
prior to dosing with Zolgensma and for about 60 days thereafter to mitigate
the chance for liver inflammation and other adverse effects. Liver function,
platelet count, and troponin-I are monitored after Zolgensma infusion. See
Zolgensma Package Insert ( 210 KB). Current pricing
is >$2 million for this 1-time dose.
210 KB). Current pricing
is >$2 million for this 1-time dose.
Risdiplam (Evrysdi) is an
oral medication given daily that works similarly to nusinersen. It was
approved by the FDA in August 2020 for children 2 months of age and older,
including adults with SMA. It should be avoided in pregnancy and may cause
problems with male fertility. Price is about $300,000 to $400,000 annually
and is given indefinitely. See EVRYSDI Prescribing Information ( 514 KB).
Which
treatment to pick is a complex question and should be discussed carefully
with the neuromuscular specialist. Data are evolving rapidly and treatment
should be tailored to the specific risks/benefits of the child. Outcome with
any of the 3 medications is better if given/started younger than 3 months of
age and with good baseline motor function. [Ramdas: 2020] Zolgensma is only available for children younger than
2, and those children must not have antibodies to the AAV9 vector. Both
risdiplam and nusinersen are approved for use in types 1, 2, and 3, although
risdiplam cannot be given to infants younger than 2 months of age.
Nusinersen must be given by lumbar puncture and risdiplam may cause birth
defects and male infertility. These factors and others must be weighed by
the family in consultation with the Neuromuscular specialist. Additionally,
families may wish to try a second medication based on outcome of the first
medication tried. [Ramdas: 2020]
Expectant management includes increased nutritional management and attention to respiratory status before and after surgery and during acute hospital visits. Temporary supplementation with nasogastric tubes or nasojejunal tubes, or peripheral or total parental nutrition, may be helpful.
Written care plans facilitate emergency careEmergency room visits for families of children with SMA are challenging because of the need for families to detail their histories and preferences, as well as the reason for their visit. Plans for what to do in case of respiratory failure should be made and revised at non-acute medical home visits. Provide families with documentation so their resuscitation desires can be shared with other clinicians.
Early postoperative ambulation helps maintain strengthRequired inactivity following major surgery (e.g., for scoliosis or hip dislocation) may precipitate the loss of ambulation. Early re-institution of ambulation in these settings can help significantly in maintaining strength.
How should common problems be managed differently in children with Spinal Muscular Atrophy?
Growth or Weight Gain
Development (Cognitive, Motor, Language, Social-Emotional)
Viral Infections
Bacterial Infections
Systems
Neurology
Specialty Collaborations & Other Services
Neuromuscular Clinics (see UT providers [3])
Pediatric Neurology (see UT providers [8])
Respiratory
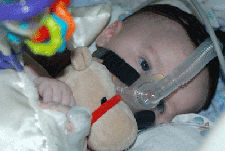
Consider a sleep study for identification of obstructive sleep apnea and central hypoventilation. These problems may occur before the child has obvious daytime problems. Nighttime BiPAP is usually necessary when the vital capacity is less than 40% of the predicted value. BiPAP can also be used in daytime during periods of increased need, such as with a respiratory illness or following surgery. The Medical Home Portal's CPAP & BIPAP Therapy for Children topic provides more information about indications for use and follow-up care.
Optimal preventive treatment includes nutritional optimization, especially with surgeries and illnesses. For some children, breath stacking methods and incentive spirometry can be taught, usually by staff at the pulmonology clinic. Breath stacking involves repetitions of taking a breath and holding it. Daily practice can be helpful for children who are losing lung capacity, but adherence is difficult.
If the child has a weak cough, percussion and postural drainage should be initiated; management of secretions is further improved by use of a cough-assist device. [Fauroux: 2008] Respiratory secretions should be managed with the help of an ENT if necessary. Consider medications to reduce secretions, botulinum toxin injections, and salivary gland ligation for children who cannot manage secretions. Swallowing problems should be managed optimally, for example, with thickened liquids and G-tube feeds; Nissen fundoplication may be helpful when reflux is present.
Ensure that immunizations, especially pneumococcal and yearly flu vaccines, are up to date. RSV prophylaxis should be given to non-sitters and most sitters. Respiratory infections and symptoms of reactive airway disease should be treated early and aggressively. If hospitalization is necessary for acute, severe respiratory illness, consider the use of non-invasive ventilation; children with SMA often have difficulty weaning from a ventilator. Supplemental oxygen without mechanical ventilation should be used with care as it may decrease respiratory drive, leading to hypercarbia and atelectasis.
Specialty Collaborations & Other Services
Pediatric Pulmonology (see UT providers [3])
Sleep Disorders (see UT providers [1])
Pediatric Otolaryngology (ENT) (see UT providers [10])
Nutrition/Growth/Bone
Poor weight gain is common and often attributed to the decreased intake of food and the increased energy demands required for the work of breathing. Registered dieticians should be involved when infants are grossly underweight. Increasing calories may involve Boosting Calories for Babies, Toddlers, and Older Children, pureeing foods, or gastrostomy feeds (either exclusively or in addition to oral feeding). Although the decision to proceed with G-tube placement can be difficult, prolonged feeding time may make the logistics of caring for non-sitters with SMA very difficult. Families may prefer to start with a nasogastric tube before proceeding with G-tube placement. For an introduction to various kinds of feeding tubes and an overview of the role of the medical home in feeding tube care, see Feeding Tubes & Gastrostomies in Children.
Excessive weight gain can also occur in sitters and walkers and can make mobility even more difficult; preventing excessive weight gain is easier than losing weight. Nutrition experts should be involved when a trend toward becoming overweight is first noted.
Specialty Collaborations & Other Services
Dieticians and Nutritionists (see UT providers [6])
Pediatric Endocrinology (see UT providers [7])
Musculoskeletal
Devices for upright positioning are prescribed for non-sitters to help with lung function, gastrointestinal function, and developmental goals. For sitters and non-sitters, standing equipment should be prescribed and used for a goal of 1 to 2 hours a day. This can help with lung and gastrointestinal function, decrease fracture risk, and delay scoliosis and contracture development. Orthotics and walking equipment should be provided, even if functional walking for all activities is not a practical goal. Prescribe scooters, wheelchairs (manual and/or powered), or other equipment, as necessary, for all children with SMA to allow participation in age-appropriate community activities. To avoid progression of contractures, implement daily range of motion exercises and early return to weight-bearing activities after surgeries.
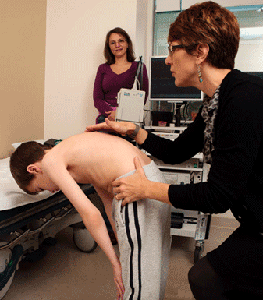
Hip subluxation and dislocation are common problems for non-sitters, sitters, and, occasionally, walkers. Imaging of the hip should be performed bi-annually or annually. Hip dislocation can make sitting balance difficult, interfere with already compromised respiratory function, and lead to chronic pain. The risks and benefits of surgery for hip subluxation should be weighed by the family who is consulting with an experienced orthopedic surgeon. When surgery is being considered for walkers, be aware that the period of inactivity may lead to contractures or changes in functional walking patterns.
Specialty Collaborations & Other Services
Pediatric Orthopedics (see UT providers [10])
Hospitals (see UT providers [57])
Pediatric Physical Medicine & Rehabilitation (see UT providers [11])
Physical Therapy (see UT providers [40])
Early Intervention for Children with Disabilities/Delays (see UT providers [51])
Nose/Throat/Mouth/Swallowing
Specialty Collaborations & Other Services
Pediatric Gastroenterology (see UT providers [2])
Pediatric General Surgery (see UT providers [2])
Gastro-Intestinal & Bowel Function
Specialty Collaborations & Other Services
Pediatric Gastroenterology (see UT providers [2])
Pediatric General Surgery (see UT providers [2])
Developmental - Behavioral Pediatrics (see UT providers [9])
Dental
Specialty Collaborations & Other Services
Pediatric Dentistry (see UT providers [50])
General Dentistry (see UT providers [93])
Recreation & Leisure
Specialty Collaborations & Other Services
Adaptive Sports (see UT providers [50])
Funding & Access to Care
Specialty Collaborations & Other Services
Neuromuscular Clinics (see UT providers [3])
Family
Appropriate care for children with SMA encompasses many options, and open discussion of alternatives are very helpful for families. Some families will choose to forego diagnostic and therapeutic interventions that they feel are invasive, whereas others will choose to proceed aggressively with interventions that might prolong life. All caregivers need to understand the family’s preferences. Discussions are best accomplished over time and in non-acute settings, rather than in the midst of an emergency room visit for pneumonia requiring intubation, for example. Preventive management, such as early implementation of non-invasive ventilation, will help avoid crises. The decision to aggressively manage SMA is a dynamic one that can be reconsidered whenever appropriate. Offer pediatric hospice in situations where aggressive intervention is not chosen.
Specialty Collaborations & Other Services
Hospice & Palliative Care (see UT providers [42])
Wish Foundations (see UT providers [18])
Medical Genetics (see UT providers [7])
Ask the Specialist
What are the earliest signs of SMA?
Signs and symptoms depend on a patient’s age and SMA subtype. Classically, children present with hypotonia, proximal weakness (shoulders and hips), feeding difficulties, and/or complications of respiratory insufficiency. Early signs of weakness include:
- Abdominal breathing or accessory muscle use
- A feeling of “slipping through hands” when held suspended by examiner under armpits
- Inability to voluntarily flex neck when supine or has head lag when pulled to sit
- In older children, difficulty rising from floor (including Gowers maneuver, full or modified)
What are the most pertinent issues to focus on during a routine well-child visit for a patient with SMA?
Ongoing and proactive assessment of sitting status, joint range of motion, feeding ability, weight, and respiratory status can enhance the quality of life and life span for children with SMA. Asking about family and community supports and access to resources that may affect the care of a child with SMA is also critical.
How does SMA affect growth and development?
Children with SMA are at risk for nutritional deficiencies and being underweight. Non-sitters, in particular, are commonly underweight secondary to decreased intake and the increased energy expenditure for the work of breathing. Standard weight charts are not applicable due to reduced muscle mass despite caloric supplementation. Children with SMA that are following the “normal” growth curves may be functionally obese and at risk for complications of metabolic syndrome.
Resources for Clinicians
On the Web
Clinical guidelines for SMA (Cure SMA)
A collection of clinical guidelines on various aspects of SMA compiled by CureSMA.
Respiratory Guidelines (Cure SMA) ( 32 KB)
Respiratory guidelines including ongoing management and equipment
Nutritional Care Guidelines (Cure SMA) ( 68 KB)
Proactive nutritional management tips for children with SMA.
Musculoskeletal issues in SMA (Cure SMA)
Compiled information about musculoskeletal issues including treatment and equipment that might be needed
Child Muscle Weakness Organization
Information and videos to help increase clinicians’ awareness of peripheral neuromuscular disease as a cause of developmental
delay in young children and the early symptoms of neuromuscular disorders; National Task Force for the Early Identification
of Childhood Neuromuscular Disorders.
Spinal Muscular Atrophy (GeneReviews)
An expert-authored, peer-reviewed, current disease description that applies genetic testing to diagnosis and management information;
U.S. National Library of Medicine.
Spinal Muscular Atrophy Type I (OMIM)
Information about clinical features, diagnosis, management, and molecular and population genetics; Online Mendelian Inheritance
in Man, authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine
Spinal Muscular Atrophy Type II (OMIM)
Information about clinical features, diagnosis, management, and molecular and population genetics; Online Mendelian Inheritance
in Man, authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine
Spinal Muscular Atrophy Type III (OMIM)
Information about clinical features, diagnosis, management, and molecular and population genetics; Online Mendelian Inheritance
in Man, authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine
Helpful Articles
PubMed search for spinal muscular atrophy in children, last 2 years.
Butterfield RJ.
Spinal Muscular Atrophy Treatments, Newborn Screening, and the Creation of a Neurogenetics Urgency.
Semin Pediatr Neurol.
2021;38:100899.
PubMed abstract / Full Text
Clinical Tools
Assessment Tools/Scales
Pediatric Evaluation of Disability Inventory (PEDI)
Helps evaluate functional disabilities of children ages 6 months to 7 years old; completion time of 45-60 minutes; manual
scoring; purchase required.
Patient Education & Instructions
Living with SMA (Cure SMA)
Excellent resource for families dealing with the day-to-day issues related to SMA. Contains information about standards of
care, school, home, equipment needs, and community resources.
Resources for Patients & Families
Information on the Web
SMA Foundation
The mission of the SMA Foundation is to accelerate treatment for children with SMA. Families will find detailed information
about SMA, up-to-date information about drugs in development, and support.
Spinal Muscular Atrophy (MedlinePlus)
A brief description of SMA, along with numerous links to further information; sponsored by the National Library of Medicine.
Genetic Conditions: Spinal Muscular Atrophy (MedlinePlus)
Excellent, detailed review of condition for patients and families; National Library of Medicine and National Institutes of
Health.
National & Local Support
Cure SMA
Offers information about research in SMA, access to support and local chapters, and opportunities for fundraising.
Rainbow Kids Palliative Care (Primary Children's Hospital)
A consultation service available to any child who is experiencing a life-threatening illness. The program helps both the child
and family deal with feelings, symptoms, and concerns during a time that may be confusing and overwhelming.
Children's Hospice International
An organization that supports the idea that critically ill children should have access to hospice/palliative care along with
curative care from the time their life-threatening illness has been diagnosed.
Never Give Up
Nonprofit organization that engages in leading scientists and policymakers to create treatments and a cure for SMA.
SMA Coalition
A coalition of SMA-focused nonprofits working toward increased awareness and help for those with SMA.
Studies/Registries
Spinal Muscular Atrophy (clinicaltrials.gov)
Studies looking at better understanding, diagnosing, and treating this condition; from the National Library of Medicine.
Services for Patients & Families in Utah (UT)
| Service Categories | # of providers* in: | UT | NW | Other states (3) (show) | | NM | NV | RI |
|---|---|---|---|---|---|---|---|---|
| Adaptive Sports | 50 | 8 | 21 | 16 | 26 | |||
| Assistive Technology Equipment | 72 | 36 | 47 | 46 | 44 | |||
| Camps for Children with Special Needs | 38 | 20 | 23 | 23 | 62 | |||
| Developmental - Behavioral Pediatrics | 9 | 1 | 2 | 3 | 12 | |||
| Dieticians and Nutritionists | 6 | 1 | 1 | 4 | 3 | |||
| Early Intervention for Children with Disabilities/Delays | 51 | 3 | 34 | 30 | 13 | |||
| General Dentistry | 93 | 1 | 12 | 13 | 47 | |||
| Hospice & Palliative Care | 42 | 3 | 5 | 26 | 4 | |||
| Hospitals | 57 | 3 | 11 | 23 | 14 | |||
| Medical Genetics | 7 | 1 | 2 | 5 | 4 | |||
| Neuromuscular Clinics | 3 | 1 | 1 | 2 | 3 | |||
| Occupational Therapy | 37 | 1 | 17 | 22 | 22 | |||
| Pediatric Dentistry | 50 | 2 | 6 | 24 | 54 | |||
| Pediatric Endocrinology | 7 | 1 | 4 | 6 | 12 | |||
| Pediatric Gastroenterology | 2 | 2 | 5 | 18 | ||||
| Pediatric General Surgery | 2 | 4 | 5 | 4 | ||||
| Pediatric Neurology | 8 | 5 | 5 | 18 | ||||
| Pediatric Orthopedics | 10 | 4 | 7 | 8 | 16 | |||
| Pediatric Otolaryngology (ENT) | 10 | 1 | 11 | 5 | 7 | |||
| Pediatric Physical Medicine & Rehabilitation | 11 | 3 | 3 | 3 | 6 | |||
| Pediatric Pulmonology | 3 | 4 | 4 | 6 | ||||
| Physical Therapy | 40 | 12 | 9 | 7 | ||||
| Rec Centers, Parks, Zoos & Museums | 61 | 2 | 9 | 38 | 22 | |||
| Sleep Disorders | 1 | 2 | ||||||
| Speech - Language Pathologists | 65 | 4 | 23 | 11 | 34 | |||
| Swallow Study | 1 | 1 | ||||||
| Wish Foundations | 18 | 14 | 14 | 16 | 17 | |||
For services not listed above, browse our Services categories or search our database.
* number of provider listings may vary by how states categorize services, whether providers are listed by organization or individual, how services are organized in the state, and other factors; Nationwide (NW) providers are generally limited to web-based services, provider locator services, and organizations that serve children from across the nation.
Bibliography
Bach JR, Saltstein K, Sinquee D, Weaver B, Komaroff E.
Long-term survival in Werdnig-Hoffmann disease.
Am J Phys Med Rehabil.
2007;86(5):339-45 quiz 346-8, 379.
PubMed abstract
Baker M, Griggs R, Byrne B, Connolly AM, Finkel R, Grajkowska L, Haidet-Phillips A, Hagerty L, Ostrander R, Orlando L, Swoboda
K, Watson M, Howell RR.
Maximizing the Benefit of Life-Saving Treatments for Pompe Disease, Spinal Muscular Atrophy, and Duchenne Muscular Dystrophy
Through Newborn Screening: Essential Steps.
JAMA Neurol.
2019.
PubMed abstract
Butterfield RJ.
Spinal Muscular Atrophy Treatments, Newborn Screening, and the Creation of a Neurogenetics Urgency.
Semin Pediatr Neurol.
2021;38:100899.
PubMed abstract / Full Text
Carré A, Empey C.
Review of Spinal Muscular Atrophy (SMA) for Prenatal and Pediatric Genetic Counselors.
J Genet Couns.
2016;25(1):32-43.
PubMed abstract
Cobben JM, Lemmink HH, Snoeck I, Barth PA, van der Lee JH, de Visser M.
Survival in SMA type I: a prospective analysis of 34 consecutive cases.
Neuromuscul Disord.
2008;18(7):541-4.
PubMed abstract
De Vivo DC, Bertini E, Swoboda KJ, Hwu WL, Crawford TO, Finkel RS, Kirschner J, Kuntz NL, Parsons JA, Ryan MM, Butterfield
RJ, Topaloglu H, Ben-Omran T, Sansone VA, Jong YJ, Shu F, Staropoli JF, Kerr D, Sandrock AW, Stebbins C, Petrillo M, Braley
G, Johnson K, Foster R, Gheuens S, Bhan I, Reyna SP, Fradette S, Farwell W.
Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results
from the Phase 2 NURTURE study.
Neuromuscul Disord.
2019;29(11):842-856.
PubMed abstract / Full Text
Fauroux B, Guillemot N, Aubertin G, Nathan N, Labit A, Clément A, Lofaso F.
Physiologic benefits of mechanical insufflation-exsufflation in children with neuromuscular diseases.
Chest.
2008;133(1):161-8.
PubMed abstract
Finkel RS, McDermott MP, Kaufmann P, Darras BT, Chung WK, Sproule DM, Kang PB, Foley AR, Yang ML, Martens WB, Oskoui M, Glanzman
AM, Flickinger J, Montes J, Dunaway S, O'Hagen J, Quigley J, Riley S, Benton M, Ryan PA, Montgomery M, Marra J, Gooch C, De
Vivo DC.
Observational study of spinal muscular atrophy type I and implications for clinical trials.
Neurology.
2014;83(9):810-7.
PubMed abstract / Full Text
Finkel RS, Mercuri E, Meyer OH, Simonds AK, Schroth MK, Graham RJ, Kirschner J, Iannaccone ST, Crawford TO, Woods S, Muntoni
F, Wirth B, Montes J, Main M, Mazzone ES, Vitale M, Snyder B, Quijano-Roy S, Bertini E, Davis RH, Qian Y, Sejersen T.
Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations;
other organ systems; and ethics.
Neuromuscul Disord.
2018;28(3):197-207.
PubMed abstract
Hamilton G, Gillingwater TH.
Spinal muscular atrophy: going beyond the motor neuron.
Trends Mol Med.
2013;19(1):40-50.
PubMed abstract
Kaindl AM, Guenther UP, Rudnik-Schöneborn S, Varon R, Zerres K, Schuelke M, Hübner C, von Au K.
Spinal muscular atrophy with respiratory distress type 1 (SMARD1).
J Child Neurol.
2008;23(2):199-204.
PubMed abstract
Ke Q, Zhao ZY, Mendell JR, Baker M, Wiley V, Kwon JM, Alfano LN, Connolly AM, Jay C, Polari H, Ciafaloni E, Qi M, Griggs RC,
Gatheridge MA.
Progress in treatment and newborn screening for Duchenne muscular dystrophy and spinal muscular atrophy.
World J Pediatr.
2019;15(3):219-225.
PubMed abstract
Khatri IA, Chaudhry US, Seikaly MG, Browne RH, Iannaccone ST.
Low bone mineral density in spinal muscular atrophy.
J Clin Neuromuscul Dis.
2008;10(1):11-7.
PubMed abstract
Kirschner J, Butoianu N, Goemans N, Haberlova J, Kostera-Pruszczyk A, Mercuri E, van der Pol WL, Quijano-Roy S, Sejersen T,
Tizzano EF, Ziegler A, Servais L, Muntoni F.
European ad-hoc consensus statement on gene replacement therapy for spinal muscular atrophy.
Eur J Paediatr Neurol.
2020;28:38-43.
PubMed abstract / Full Text
Kolb SJ, Battle DJ, Dreyfuss G.
Molecular functions of the SMN complex.
J Child Neurol.
2007;22(8):990-4.
PubMed abstract
Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J, Main M, Mazzone ES, Vitale M, Snyder B, Quijano-Roy S, Bertini E, Davis
RH, Meyer OH, Simonds AK, Schroth MK, Graham RJ, Kirschner J, Iannaccone ST, Crawford TO, Woods S, Qian Y, Sejersen T.
Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and
nutritional care.
Neuromuscul Disord.
2018;28(2):103-115.
PubMed abstract
Messina S, Pane M, De Rose P, Vasta I, Sorleti D, Aloysius A, Sciarra F, Mangiola F, Kinali M, Bertini E, Mercuri E.
Feeding problems and malnutrition in spinal muscular atrophy type II.
Neuromuscul Disord.
2008;18(5):389-93.
PubMed abstract
Michelson D, Ciafaloni E, Ashwal S, Lewis E, Narayanaswami P, Oskoui M, Armstrong MJ.
Evidence in focus: Nusinersen use in spinal muscular atrophy: Report of the Guideline Development, Dissemination, and Implementation
Subcommittee of the American Academy of Neurology.
Neurology.
2018;91(20):923-933.
PubMed abstract
Parente V, Corti S.
Advances in spinal muscular atrophy therapeutics.
Ther Adv Neurol Disord.
2018;11:1756285618754501.
PubMed abstract / Full Text
Prior TW.
Carrier screening for spinal muscular atrophy.
Genet Med.
2008;10(11):840-2.
PubMed abstract
Ramdas S, Servais L.
New treatments in spinal muscular atrophy: an overview of currently available data.
Expert Opin Pharmacother.
2020;21(3):307-315.
PubMed abstract
Ross LF, Kwon JM.
Spinal Muscular Atrophy: Past, Present, and Future.
Neoreviews.
2019;20(8):e437-e451.
PubMed abstract
Schroth MK.
Special considerations in the respiratory management of spinal muscular atrophy.
Pediatrics.
2009;123 Suppl 4:S245-9.
PubMed abstract
Shanmugarajan S, Swoboda KJ, Iannaccone ST, Ries WL, Maria BL, Reddy SV.
Congenital bone fractures in spinal muscular atrophy: functional role for SMN protein in bone remodeling.
J Child Neurol.
2007;22(8):967-73.
PubMed abstract
Sumner CJ.
Molecular mechanisms of spinal muscular atrophy.
J Child Neurol.
2007;22(8):979-89.
PubMed abstract
Verhaart IEC, Robertson A, Wilson IJ, Aartsma-Rus A, Cameron S, Jones CC, Cook SF, Lochmüller H.
Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review.
Orphanet J Rare Dis.
2017;12(1):124.
PubMed abstract / Full Text
Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, Aloysius A, Morrison L, Main M, Crawford TO, Trela A.
Consensus statement for standard of care in spinal muscular atrophy.
J Child Neurol.
2007;22(8):1027-49.
PubMed abstract