
 Get More Help in Utah
Get More Help in UtahMucopolysaccharidosis Type I (MPS 1)
Overview

Clinical variability among the mucopolysaccharidoses is broad. Historically, the most severe form of MPS I was classified as Hurler syndrome, whereas the intermediate form was called Hurler-Scheie syndrome, and the least severe form was called Scheie syndrome. More recently, the terms "severe" MPS I for Hurler syndrome and "attenuated" MPS for Hurler-Scheie or Scheie syndromes are preferred.
All individuals with MPS I require an extensive multidisciplinary team approach to care given the significant multi-organ system involvement associated with this condition.
Other Names & Coding
E76.0, Mucopolysaccharidosis, type I
E76.01, Hurler's syndrome
E76.02, Hurler-Scheie syndrome
E76.03, Scheie syndrome
For further coding details, see ICD-10 for Disorders of the Glycosaminoglycan Metabolism (icd10data.com).
Prevalence
Genetics
Prognosis
Practice Guidelines
The following presents a consensus on the management and treatment of individuals with MPS I.
Muenzer J, Wraith JE, Clarke LA.
Mucopolysaccharidosis I: management and treatment guidelines.
Pediatrics.
2009;123(1):19-29.
PubMed abstract / Full Text
Roles of the Medical Home
Once a diagnosis is established, coordination of selected MPS I therapies should be initiated by the medical home. In those with severe MPS I, establishing a collaboration with the transplant team is imperative for long-term care. For those with attenuated MPS I who elect to have ERT, initial infusions should be carried out in a hospital-based unit familiar with potential adverse events of enzyme replacement therapy. After at least 8 infusions and the placement of an indwelling catheter, the medical home can establish a relationship with the pharmaceutical company that supplies the recombinant enzyme and develop a plan with the metabolic genetics team and home health nursing to provide infusions in the home.
Given the rapid progression of the disease, assessments several times a year are indicated for children with severe MPS I. Those with the attenuated forms may also benefit from frequent visits, though the need will depend upon their clinical status. Because of the extensive and widespread organ system involvement, every child with MPS I should receive multidisciplinary and subspecialty care from a center with expertise in lysosomal storage disorders.
Clinical Assessment
Overview
 344 KB) for a recommended schedule of assessments.
344 KB) for a recommended schedule of assessments.
Pearls & Alerts for Assessment
Elevation of urinary glycosaminoglycans (GAGs) is not specific to MPS IDefinitive diagnosis of MPS I requires demonstration of deficient activity of the lysosomal enzyme α-L-iduronidase in peripheral blood leukocytes or cultured fibroblasts.
Anesthesia in individuals with MPS I is very riskyAirway management during anesthesia in individuals with MPS I is difficult and should be performed by anesthesiologists aware of the significant risks associated with sedation, such as airway compromise and cervical joint limitation secondary to GAG accumulation. In treated patients, these high risks abate somewhat with decreased accumulation of airway GAGs.
Signs of hydrocephalus/intracranial pressure may be subtleA rapid increase in head circumference, acute behavioral changes, or headaches could be signs of hydrocephalus. If they are present, an ophthalmologic assessment of the optic nerve, brain MRI, and lumbar puncture with measurement of opening pressure of cerebrospinal fluid (CSF) are helpful in assessing the degree of pressure elevation. On MRI, the ventricles can be resistant to volume changes with increased pressure due to GAG accumulation in the intracranial arachnoid and meningeal membranes.
Carpal tunnel syndromeFinger tingling and pain with children chewing on fingertips could represent carpal tunnel syndrome with median nerve entrapment.
Risk for spinal cord compressionBecause of the risk for ligamentous laxity and vertebral hypoplasia leading to spinal cord compression, parents/patients should be instructed to avoid "high-risk" activities, such as contact sports, and should be cautioned about manipulation of the cervical spine.
Screening
For the Condition
Newborn screening has been implemented in a number of states, including Utah in 2019, and is under consideration in other states where it will likely be added to the Newborn Screen Panel.
Of Family Members
Carrier screening of parents and other family members via enzyme analysis is not recommended since there can be significant overlap in enzyme levels between carriers and non-carriers. Carrier screening via molecular genetic testing is indicated in family members if the mutations within the family are known.
Presentations
Manifestations observed include:
- Frequent upper airway infections with otitis media
- Hepatosplenomegaly
- Corneal clouding
- Cardiac involvement, valve disease
- Skeletal dysplasia (dysostosis multiplex)
- Growth delay
- Profound neurological involvement (in the severe form only)
- Macrocephaly
Orthopedic manifestations cause significant morbidity and discomfort to individuals with mucopolysaccharidosis type I (MPS I). Orthopedic concerns include:
- Progressive arthropathy leading to severe joint deformity (occurs in all patients)
- Early bone involvement, with diminution of linear growth by age 3 in children with severe MPS I
- Carpal tunnel syndrome is common, but early symptoms (pain, tingling, and numbness) often go unreported. More commonly, parents notice increased difficulty with fine motor skills (a late symptom) or gnawing on fingertips.
Individuals with attenuated MPS I may have severe progressive bone involvement despite absence of cognitive impairment. Joint stiffness and contractures may present initially in the fingers resulting in the characteristic "claw hand." Progressive skeletal dysplasia, or dysostosis multiplex, is seen in all individuals with severe MPS I. Dysostosis multiplex congenita is a radiologic finding with a number of related features, including:
- Dolichocephaly
- Spatulate ribs (oar-like)
- Wide diaphyses and narrow epiphyses
- Wide metacarpals, phalanges, metatarsals (bullet-like)
- Hypoplastic acetabulae
- Kyphoscoliosis (gibbus deformity)
- Anterior beaking of vertebrae
- Platyspondyly
- Ossification defects of the lower vertebral bodies
Complications may include:
- Spinal nerve entrapment, including sciatica
- Acute spinal injury
- Atlanto-occipital instability
- Knees that are prone to valgus and varus deformities
- Pelvic abnormalities
- Carpal tunnel syndrome
- Progressive and debilitating hip deformity leading to early hip replacement
- Resultant joint pain
Diagnostic Criteria
- Confirming deficiency of α-L-iduronidase in peripheral blood leukocytes or cultured fibroblasts
- Molecular genetic testing for mutations in IDUA, the gene that encodes α-L-iduronidase
Clinical Classification
Severe MPS I (formerly Hurler syndrome): Infants may appear normal at birth. Coarsening of facial features and enlargement of the liver and spleen occurs over the first few months. If untreated, death occurs in the first decade. The following features are usually present at an early age:
- Skeletal deformities including gibbus and distinctive radiographic findings
- Obstructive airway disease
- Progressive and severe physical problems
- Communicating hydrocephalus
- Limited language ability and progressive mental deterioration
- Hepatosplenomegaly
- Recurrent ear/nose infections
- Multiple hernias; umbilical and inguinal
- Coarse facial features with increased body hair for family background
- Dysostosis multiplex
- Arthropathy
- Corneal clouding
- Joint stiffness
- Hearing loss
- Cardiac involvement, including valvular heart disease
- Spinal cord compression
- Obstructive airway disease
- Corneal clouding
- Sleep apnea
- Arthropathy and joint stiffness
- Carpal tunnel syndrome and other nerve entrapment (such as cervical cord compression)
- Valvular heart disease
- Visual impairment
- Mild facial coarseness
- Spinal cord compression
Differential Diagnosis
History & Examination
344 KB) for a recommended schedule of assessments.
Current & Past Medical History
- 0 - 6 months
- Chronic rhinitis
- Recurrent otitis media or "glue ear" with thick cerumen
- Umbilical or inguinal hernia
- Above normal growth and head size
- Gibbus of the thoracolumbar spine
- 6 months -12 years
- Distinctive facial gestalt, with coarsening over time
- Hepatosplenomegaly
- Skeletal deformities
- Joint stiffness - especially distal interphalangeal and elbow joints
- Developmental delay
- Corneal clouding
- Chronic rhinitis
- Recurrent otitis media or "glue ear"
- 12+ years
- Corneal clouding
- Joint stiffness
- Valvular heart disease
- Cardiopulmonary disease with easy fatigue
Family History
- Are there any individuals with developmental delays, regression, or learning problems?
- Any children who died at a young age?
- Any individuals with joint problems? Pain, tingling, or numbness in the hands or difficulty with fine motor skills? (It may signify carpal tunnel syndrome.)
- Any individuals with heart problems? Recurrent ear infections or chronic rhinitis?
- Which ethnicities exist in the family?
- Are the parents related to each other (consanguinity)?
Developmental & Educational Progress
Social & Family Functioning
Physical Exam
A physical exam is recommended every 6 months. See Table 1 (page 3) in Mucopolysaccharidosis I: Management and Treatment Guidelines (AAP) ( 344 KB) for a recommended schedule of assessments.
General
Vital Signs
Growth Parameters
Children with severe MPS I may have above-normal growth and head circumference in the first 6 months of life; however, look for growth to plateau and diminution of linear growth by 3 years. Assess weight and length/height.
Skin
HEENT/Oral
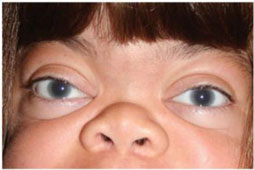
Corneal clouding (image, left) occurs in all individuals
with MPS I, and its presence should be documented.
Heart
Stenotic or regurgitant murmurs are common, secondary to GAG accumulation in cardiac valves (primarily aortic and mitral valves). Check for tachycardia, which may be present due to aberrant conduction pathways. Absence of a murmur on physical exam does not exclude cardiac involvement. Echocardiograms are needed to assess valve stenosis, regurgitation, and ventricular function.
Abdomen
Protruding abdomen and inguinal or umbilical hernias may be present. Hepatosplenomegaly is common in children and adolescents.
Extremities/Musculoskeletal
Contractures of small and large joints and gibbus deformity of the spine are very common in the early stages of MPS I. Assess for joint stiffness (elbows with lack of supination/pronation range of motion, shoulders with inability to fully extend arms over the head, hand limitations, and contractures (initially present in the fingers resulting in the characteristic "claw-hand"); limited hip abduction and genu valgum. Assess grip strength and thenar muscle atrophy, related to carpal tunnel syndrome.
Neurologic Exam
Progressive compression of the spinal cord with resulting cervical myelopathy is relatively common in older individuals. Communicating hydrocephalus may occur without ventriculomegaly, due to the thickened arachnoid membrane. Presence of acute behavioral changes, headaches, and vomiting should trigger an evaluation of hydrocephalus.
Testing
Sensory Testing
- Hearing and speech discrimination
- Visual acuity, including peripheral vision
- Fingertip touch
Laboratory Testing
Imaging
Genetic Testing
Other Testing
- Annual ophthalmology assessments should include measurement of visual acuity and intraocular pressure, slit lamp examination of the cornea, and electroretinography. Assessment of optic nerve for signs of intracranial pressure
- Regular sleep assessments, with sleep studies as needed, to identify the presence of sleep apnea, both obstructive and central
- Pulmonary function tests when of age with regular assessments
- Dental exam every 6-12 months
- Annual audiology exam to determine the degree and cause of hearing impairment
Specialty Collaborations & Other Services
Medical Genetics (see UT providers [7])
Genetic Testing and Counseling (see UT providers [11])
Early Intervention for Children with Disabilities/Delays (see UT providers [51])
Pediatric Ophthalmology (see UT providers [4])
Pediatric Orthopedics (see UT providers [10])
Pediatric Cardiology (see UT providers [4])
Audiology (see UT providers [22])
Pediatric Neurology (see UT providers [8])
Pediatric Otolaryngology (ENT) (see UT providers [10])
Pediatric Pulmonology (see UT providers [3])
Pediatric Physical Medicine & Rehabilitation (see UT providers [11])
Developmental - Behavioral Pediatrics (see UT providers [9])
Pediatric Gastroenterology (see UT providers [2])
Pediatric General Surgery (see UT providers [2])
Wish Foundations (see UT providers [18])
Treatment & Management
Overview
- Hematopoietic stem cell transplantation (HSCT) for severe MPS I increases survival, reduces facial coarseness and hepatosplenomegaly, improves hearing, and preserves normal heart function. If HSCT is accomplished before significant developmental delay (usually before 2 years old), the degree and rate of cognitive decline will likely be reduced; however, individuals may still have mild intellectual disability. Cardiac valvular disease, skeletal manifestations, and corneal clouding may continue to progress. [Kunin-Batson: 2016]
- Enzyme replacement therapy (ERT) can significantly reduce liver size, improve the distance traveled in a 6-minute walk test, decrease joint restriction, lessen sleep apnea, and improve breathing in individuals with attenuated MPS I. The benefit of ERT in those with severe disease has not been assessed; 1 patient with severe MPS who had been treated for 3 years with ERT continued to experience a decline in respiratory status, musculoskeletal and spinal involvement, and developmental skills. [Thomas: 2006] Because the recombinant enzyme is not thought to cross the blood-brain barrier, the best option to reduce the risk of cognitive impairment remains early HSCT.
- Combined HSCT and ERT is another option for individuals with severe MPS I. Providing ERT before and into the peri-HSCT period may improve the child's existing respiratory and cardiac manifestations to an extent that may reduce the risk of transplant-related complications. [Tolar: 2008] See Mucopolysaccharidosis Type I (MPS 1) Hematopoietic Stem Cell Transplantation & Enzyme Replacement Therapy for details about therapy and dosing regimen.
- Under investigation for the treatment of cognitive decline in severe MPS I is intrathecal ERT (injecting the enzyme into the spinal fluid). Some long-term benefits to the heart from successful bone marrow transplantation have been documented. [Braunlin: 2003]
Pearls & Alerts for Treatment & Management
Spine instabilityDysostosis multiplex can lead to instability of the spine, including the atlantoaxial joint. Careful positioning and avoidance of hyperextension of the neck are necessary.
Narrow tracheaInduction of anesthesia can be difficult; smaller than anticipated endotracheal tubes are usually required for intubation because the trachea may be narrowed and the vocal cords thickened.
Postoperative recovery issuesRecovery from anesthesia may be slow and postoperative airway obstruction is a common problem. Overnight stays for typical outpatient procedures are prudent.
How should common problems be managed differently in children with Mucopolysaccharidosis Type I (MPS 1)?
Growth or Weight Gain
Bacterial Infections
Over the Counter Medications
Systems
Cardiology
- Thickening of the mitral and aortic valves leading to regurgitation and/or stenosis
- Myocardial thickening that contributes to heart failure
- Cardiac arrhythmias that lead to heart block, resulting in the need for a permanent pacemaker
- Narrowing of the coronary arteries, often within the first year of life - complete occlusions of the coronary arteries have been reported within the first 5 years of life, with sudden death occurring during illness or with blood pressure changes associated with anesthesia
- Narrowing of the thoracic aorta, possibly resulting in elevated blood pressure in the head and neck with normal blood pressure in the legs
- Congenital cardiac abnormalities, including atrial septal defect or patent ductus arteriosus
- Cor pulmonale
Specialty Collaborations & Other Services
Pediatric Cardiology (see UT providers [4])
Musculoskeletal
While orthopedic surgery may greatly enhance the quality of life, there are significant risks for MPS I patients who undergo general anesthesia. Procedures that require general anesthesia should be conducted in an experienced center that is aware of the risks in this population:
- Dysostosis multiplex can lead to instability of the spine, including the atlantoaxial joint. Careful positioning and avoidance of hyperextension of the neck are necessary.
- Maintaining an adequate airway for induction of anesthesia can be difficult.
- Smaller than anticipated endotracheal tubes are often required because the trachea may be narrowed and the vocal cords thickened.
Spinal cord compression or instability of the neck may result in myelopathy (and rarely quadriparesis). Patients should be instructed to avoid "high-risk" activities, such as contact sports, and parents should be cautioned about manipulation of the cervical spine. Neurosurgical stabilization of the spine may be indicated for those with signs of cord compression.
Thoracolumbar kyphosis (gibbus deformity), due to poor bone growth in the anterior vertebrae, occurs in about 90% of patients with severe MPS I and is the most common orthopedic abnormality. Prior to hematopoietic stem cell transplant, surgery for gibbus deformities was not performed on patients with MPS I due to their limited life expectancy. Following hematopoietic stem cell transplant, kyphosis will progress in about a third of patients and surgery will be required before age 9; in another third, it will not progress and surgery may be required later; kyphosis will improve in the remaining patients.
Myelopathy and respiratory problems can occur later in life if kyphosis is left untreated. Bracing may slow the progression of kyphosis and scoliosis, delaying but not preventing surgery. However, bracing is often not tolerated by young children and generally not recommended.
The presence of myelopathy is also an indication for surgery.
- Surgery for kyphosis almost always requires incisions from the front and the back.
- Metal hardware is typically stainless steel or titanium and is generally not removed unless there are complications.
- Most children will require some combination of a cast or brace for between 3 months and a year.
- An unsuccessful fusion can be painful and require repeat surgery.
Scoliosis may accompany kyphosis. When left untreated, scoliosis can lead to difficulty expanding the chest wall. Delaying spinal surgery may allow for maximal growth of the spine and further development of already dysplastic bone.
Hip dysplasia, to some degree, is found in nearly all children with severe MPS I. It is not responsive to hematopoietic stem cell transplant or enzyme replacement therapy; the majority of children eventually require corrective surgery. Without hip surgery, there is progressive pain and stiffness and eventually frank dislocation of the hips with a dramatic reduction in walking ability. Surgery involves repositioning the bones and tightening the soft tissues around the hip.
Genu valgum (knock-knees) severe enough to require surgery occurs in about 50% of children with severe MPS I post-transplant. The indication for surgery is a knee deformity greater than 15 degrees. Children with MPS I also suffer from stiff knees, which prevent full extension and result in a crouched gait. Physical therapy can optimize knee motion and enhance walking. Knee staples are placed in the bone on the inner side of the knee to prevent bone growth on the inner side to allow the outer side to catch up. Occasionally, the staples dislodge and may need to be removed and replaced. In small children, staples will not work and osteotomies in large bones around the knees may be required.
Carpal tunnel syndrome, trigger digits, and contractures of the fingers are common. Since there is a high incidence of undetected carpal tunnel syndrome, close monitoring is recommended, including regular evaluation by an occupational therapist preferably specialized in hands. Surgery consists of releasing the constricting tissue over the median nerve and removing the deposits on the surrounding tendons. Trigger digits are treated by opening 1 or 2 of the many pulleys in each finger and cleaning the tendons of GAG material. These surgeries are relatively minor but may be beneficial.
Specialty Collaborations & Other Services
Pediatric Orthopedics (see UT providers [10])
Respiratory
Airway obstruction and sleep apnea are common. Enlarged tonsils and adenoids may need to be removed. Supplemental oxygen and/or a tracheostomy is sometimes necessary for patients with chronic dyspnea. For sleep apnea, CPAP & BIPAP Therapy for Children are non-invasive options if the patient can adjust to wearing the mask and mouthpiece at night. Complications include the buildup of mucus in the nasal passages or discomfort if the pressure is inappropriately high. Pulmonary function testing with lung vital capacity measurements are helpful in monitoring cardio-respiratory status and response to therapies.
Specialty Collaborations & Other Services
Pediatric Pulmonology (see UT providers [3])
Neurology
Prior to surgery, the cervical region should be evaluated for evidence of narrowing at the foramen magnum, which places the spinal cord at risk. A laminectomy may need to be performed at the time of surgery. Consider a high-pressure shunt (10-15 mm Hg) to prevent rapid decompression.
Specialty Collaborations & Other Services
Pediatric Neurology (see UT providers [8])
Pediatric Neurosurgery (see UT providers [2])
Ears/Hearing
Otitis media is a persistent problem in children with MPS I. Pseudomonas aeruginosa and Staphylococcus aureus are more common as children age. Pneumococcal vaccine and corticosteroids may be helpful. Some children may benefit from eliminating common food allergens (soy, citrus, peanuts, wheat, fish, eggs, corn, and tomatoes) from their diet. With draining ears, fungal infection should be considered. Tympanostomy tubes with heavy-duty grommets are recommended; removal of the adenoids and tonsils are often necessary to prevent recurrent and persistent infections.
Specialty Collaborations & Other Services
Audiology (see UT providers [22])
Pediatric Otolaryngology (ENT) (see UT providers [10])
Speech and Hearing Services (see UT providers [29])
Eyes/Vision
Specialty Collaborations & Other Services
Pediatric Ophthalmology (see UT providers [4])
Gastro-Intestinal & Bowel Function
Diarrhea, possibly due to a defect in the autonomic nervous system, is common in younger patients. Try decreasing roughage in the diet and, if taking antibiotics, taking a live-culture yogurt or probiotics. Constipation can be a problem in older patients, requiring laxatives or enemas. Information for management of constipation can be found in the Portal's Constipation.
Specialty Collaborations & Other Services
Pediatric Gastroenterology (see UT providers [2])
Dental
Specialty Collaborations & Other Services
General Dentistry (see UT providers [93])
Development (general)
Individuals with the attenuated forms of MPS I may have normal to mildly delayed development, and intelligence can range from normal cognition to mild learning disabilities. Baseline cognition and mental status should be carefully assessed; acute changes in cognition or mental status should be promptly evaluated as this may represent increased intracranial pressure due to hydrocephalus. Families may need assistance in developing plans for school (IEP and 504). Information can be found at School Accommodations: IEPs & 504s.
Specialty Collaborations & Other Services
Developmental - Behavioral Pediatrics (see UT providers [9])
Ask the Specialist
What are the first clinical indications that a child could have MPS I?
For severe MPS I, children may have recurrent ear infections, umbilical or inguinal hernias, gibbus, above-average growth and large head circumference, and coarsening of facial features with excess body hair. Slowing of growth, increased joint stiffness, hepatosplenomegaly, and developmental delays may be noted shortly thereafter.
What treatments are available for MPS I?
Currently, there are 2 FDA-approved treatment options for MPS I. 1) Hematopoietic stem cell transplantation (HSCT) and 2) Enzyme replacement therapy (ERT). See Mucopolysaccharidosis Type I (MPS 1) for details.
Resources for Clinicians
On the Web
Mucopolysaccharidosis Type I (GeneReviews)
Detailed information addressing clinical characteristics, diagnosis/testing, management, genetic counseling, and molecular
pathogenesis; from the University of Washington and the National Library of Medicine.
Hurler Syndrome (OMIM)
Information about clinical features, diagnosis, management, and molecular and population genetics; Online Mendelian Inheritance
in Man, authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine
MPS 1 - Information for Health Care Professionals (Genzyme)
Clinical information about diagnosis and genetics; Genzyme Therapeutics, manufacturer of Aldurazyme (laronidase) enzyme replacement
therapy.
Helpful Articles
PubMed search for mucopolysaccharidosis type I in children, last 4 years.
Muenzer J.
The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations.
J Pediatr.
2004;144(5 Suppl):S27-34.
PubMed abstract
Beck M, Arn P, Giugliani R, Muenzer J, Okuyama T, Taylor J, Fallet S.
The natural history of MPS I: global perspectives from the MPS I Registry.
Genet Med.
2014;16(10):759-65.
PubMed abstract / Full Text
de Ru MH, Boelens JJ, Das AM, Jones SA, van der Lee JH, Mahlaoui N, Mengel E, Offringa M, O'Meara A, Parini R, Rovelli A,
Sykora KW, Valayannopoulos V, Vellodi A, Wynn RF, Wijburg FA.
Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis
type I: results of a European consensus procedure.
Orphanet J Rare Dis.
2011;6:55.
PubMed abstract / Full Text
Kunin-Batson AS, Shapiro EG, Rudser KD, Lavery CA, Bjoraker KJ, Jones SA, Wynn RF, Vellodi A, Tolar J, Orchard PJ, Wraith
JE.
Long-Term Cognitive and Functional Outcomes in Children with Mucopolysaccharidosis (MPS)-IH (Hurler Syndrome) Treated with
Hematopoietic Cell Transplantation.
JIMD Rep.
2016.
PubMed abstract / Full Text
Clinical Tools
Medication Guides
Aldurazyme Information for Physicians (Genzyme) ( 247 KB)
Fourteen-page handout for physicians covering pharmacology, dosage, precautions, and contraindications; Genzyme Therapeutics
(manufacturer of Aldurazyme (laronidase) enzyme replacement therapy).
Patient Education & Instructions
MPS Fact Sheets (National MPS Society)
More than 25 fact sheets about mucopolysaccharidoses and related diseases for patients, families, and providers; topics include
cardiac problems, caregiver support, family coping strategies, melatonin, transplants, pamidronate, tube feedings, stem cell
transplants, and more.
Understanding MPS (National MPS Society)
Booklets for each type of MPS with in-depth information about related physical and emotional issues; available in English
and Spanish.
Resources for Patients & Families
Information on the Web
Mucopolysaccharidosis Type I (MedlinePlus)
Information for families that includes description, frequency, causes, inheritance, other names, and additional resources;
from the National Library of Medicine.
MPS I - Information for Families (Genzyme)
Information about treatment, clinical trials, support groups, and resources related to MPS 1. Discusses the use of Aldurazyme
(laronidase) enzyme replacement therapy manufactured by Genzyme.
About Lysosomal Diseases (LDNZ)
Offers background information, family stories, newsletters, and research updates; Lysosomal Diseases New Zealand.
National & Local Support
National MPS Society
Provides detailed information, research, and support for families.
Studies/Registries
Children and Adolescents with MPS I (ClinicalTrials.gov)
Studies looking at better understanding, diagnosing, and treating this condition; from the National Library of Medicine.
Services for Patients & Families in Utah (UT)
| Service Categories | # of providers* in: | UT | NW | Other states (3) (show) | | NM | NV | RI |
|---|---|---|---|---|---|---|---|---|
| Audiology | 22 | 3 | 22 | 8 | 24 | |||
| Developmental - Behavioral Pediatrics | 9 | 1 | 2 | 3 | 12 | |||
| Early Intervention for Children with Disabilities/Delays | 51 | 3 | 34 | 30 | 13 | |||
| General Dentistry | 93 | 1 | 12 | 13 | 47 | |||
| Genetic Testing and Counseling | 11 | 6 | 6 | 12 | 8 | |||
| Medical Genetics | 7 | 1 | 2 | 5 | 4 | |||
| Pediatric Cardiology | 4 | 3 | 4 | 17 | ||||
| Pediatric Gastroenterology | 2 | 2 | 5 | 18 | ||||
| Pediatric General Surgery | 2 | 4 | 5 | 4 | ||||
| Pediatric Neurology | 8 | 5 | 5 | 18 | ||||
| Pediatric Neurosurgery | 2 | 1 | 2 | 4 | 3 | |||
| Pediatric Ophthalmology | 4 | 1 | 6 | 6 | 8 | |||
| Pediatric Orthopedics | 10 | 4 | 7 | 8 | 16 | |||
| Pediatric Otolaryngology (ENT) | 10 | 1 | 11 | 5 | 7 | |||
| Pediatric Physical Medicine & Rehabilitation | 11 | 3 | 3 | 3 | 6 | |||
| Pediatric Pulmonology | 3 | 4 | 4 | 6 | ||||
| Speech and Hearing Services | 29 | 7 | 36 | 12 | 20 | |||
| Wish Foundations | 18 | 14 | 14 | 16 | 17 | |||
For services not listed above, browse our Services categories or search our database.
* number of provider listings may vary by how states categorize services, whether providers are listed by organization or individual, how services are organized in the state, and other factors; Nationwide (NW) providers are generally limited to web-based services, provider locator services, and organizations that serve children from across the nation.
Bibliography
Beck M, Arn P, Giugliani R, Muenzer J, Okuyama T, Taylor J, Fallet S.
The natural history of MPS I: global perspectives from the MPS I Registry.
Genet Med.
2014;16(10):759-65.
PubMed abstract / Full Text
Braunlin EA, Stauffer NR, Peters CH, Bass JL, Berry JM, Hopwood JJ, Krivit W.
Usefulness of bone marrow transplantation in the Hurler syndrome.
Am J Cardiol.
2003;92(7):882-6.
PubMed abstract
de Ru MH, Boelens JJ, Das AM, Jones SA, van der Lee JH, Mahlaoui N, Mengel E, Offringa M, O'Meara A, Parini R, Rovelli A,
Sykora KW, Valayannopoulos V, Vellodi A, Wynn RF, Wijburg FA.
Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis
type I: results of a European consensus procedure.
Orphanet J Rare Dis.
2011;6:55.
PubMed abstract / Full Text
Kunin-Batson AS, Shapiro EG, Rudser KD, Lavery CA, Bjoraker KJ, Jones SA, Wynn RF, Vellodi A, Tolar J, Orchard PJ, Wraith
JE.
Long-Term Cognitive and Functional Outcomes in Children with Mucopolysaccharidosis (MPS)-IH (Hurler Syndrome) Treated with
Hematopoietic Cell Transplantation.
JIMD Rep.
2016.
PubMed abstract / Full Text
Moore D, Connock MJ, Wraith E, Lavery C.
The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK.
Orphanet J Rare Dis.
2008;3:24.
PubMed abstract / Full Text
Muenzer J.
The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations.
J Pediatr.
2004;144(5 Suppl):S27-34.
PubMed abstract
Muenzer J, Wraith JE, Clarke LA.
Mucopolysaccharidosis I: management and treatment guidelines.
Pediatrics.
2009;123(1):19-29.
PubMed abstract / Full Text
Thomas JA, Jacobs S, Kierstein J, Van Hove J.
Outcome after three years of laronidase enzyme replacement therapy in a patient with Hurler syndrome.
J Inherit Metab Dis.
2006;29(6):762.
PubMed abstract
Tolar J, Grewal SS, Bjoraker KJ, Whitley CB, Shapiro EG, Charnas L, Orchard PJ.
Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome.
Bone Marrow Transplant.
2008;41(6):531-5.
PubMed abstract / Full Text